BatMass: mass spectrometry data visualization
BatMass is a mass-spectrometry data visualization tool, with focus on being fast and interactive. Watch a short demo of BatMass in action. To understand what it can do.
Features
-
Support for the open standard mzML and mzXML mass spectrometry data types. We are hoping to bring native vendor format support as well.
-
Viewer synchronization. Link any number of viewers and zooming/panning will be synchronized across them. If you're viewing MS1 data in one view and MS2 data in the other the retention time is synchronized, while m/z is not. Open a detected LC/MS feature table or a peptide identification table, a double click on the row will open the corresponding spectrum, or bring you to the corresponding location in a 2D Map viewer.
-
Data access layer. For the Java developers out there, the highly optimized mzML/mzXML parsers can be used in any standalone Java program as a simple jar dependency. Parsing has been manually tuned to produce few garbage objects, thus minimizing time spent in GC (Garbage Collection), the speed is comparable to or better than in C/C++ implementations. The API for LC/MS data files gives access to most of the features supported by mzML/mzXML standards.
See the getting started guide for instructions how to get it up and running.
Some Examples
The layout of windows is free and customizable by dragging. 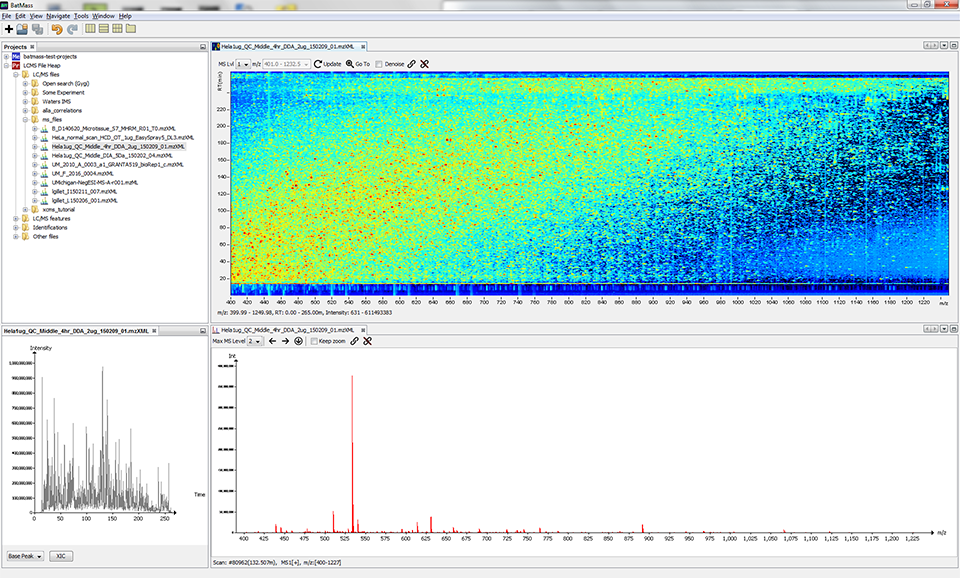
Compare multiple experiments at once. The bottom-middle run in this figure is a
blank (no sample was injected), while the other 5 were runs with some sample.
Amazing how much stuff comes from the background. 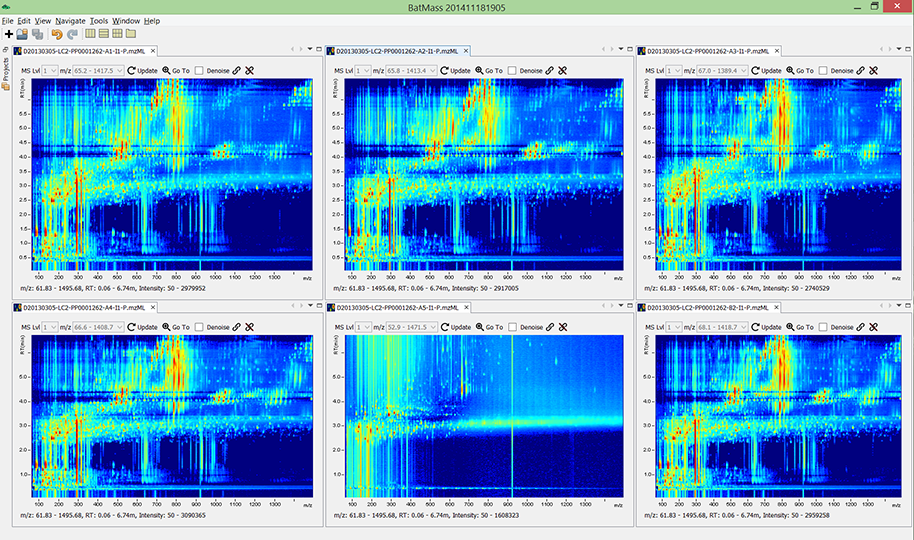
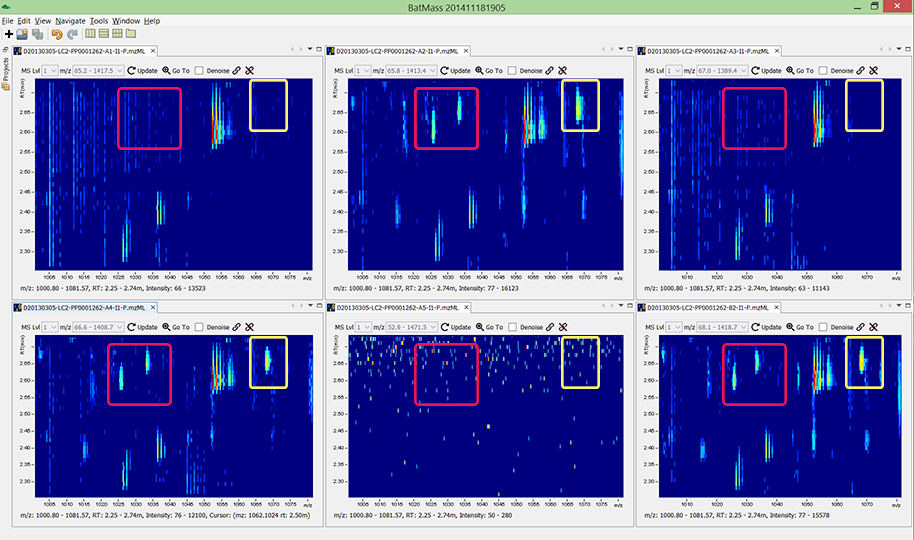
And here are the same runs but zoomed in to a small region of m/z and retentino
time. Look at the color-marked regions. Let's just accept that zero-values are a
thing, there is no need to try extracting noise to do gap-filling in data.
About BatMass
BatMass is a mass-spectrometry data visualization tool, with focus on
being fast and interactive while providing comprehensive visualizations without
any parameter tweaking. It is written in pure Java and built on top of the
NetBeans Platform.
BatMass development is kindly supported by YourKit Java Profiler
Citing / Referencing the work
If you use BatMass for your research or work, please cite the following paper:
Avtonomov D.M. et al: J. Proteome Res. June 16, 2016. DOI:
10.1021/acs.jproteome.6b00021.
Contacts
The author and maintainer of the project
Dmitry Avtonomov, Ph.D.
University of Michigan, Ann Arbor
Email: dmitriya@umich.edu
General inquiries
Alexey Nesvizhskii, Ph.D.
University of Michigan, Ann Arbor
Email: nesvi@umich.edu
http://www.nesvilab.org
Please use the bug tracker to ask questions, submit feature requests and bug reports.
This software was made possible thanks to:
 |
YourKit Java Profiler |
 |
Intellij IDEA IDE |
| NetBeans Platform community |